比较BTK抑制剂的配体与蛋白静电势
摘要
Flare[1]是Cresset的基于结构药物设计软件,在本文中它内置的蛋白相互作用势分析方法用于计算BTK激酶活性位点里精细的静电特征。相互作用势映射用于比较几个精选的BTK配体以详尽地了解配体结合的构效关系(SAR)。FLARE里的3D-RISM分析也用来研究了BTK活性位点里结晶水分子的分布稳定性。
前言
Bruton酪氨酸激酶是非受体酪氨酸激酶Tec家族的成员。最近的文献发现[2]表明Btk抑制可能是治疗自身免疫性疾病如类风湿性关节炎的有吸引力的方法,类风湿性关节炎是一种以关节肿胀和侵蚀为特征的进行性自身免疫性疾病。
已发表的X射线晶体结构PDB:4ZLZ显示4RV配体通过在铰链区与Glu475和Met477形成H键相互作用而与Btk的活性位点相互作用(图1-左)。吡啶基环与Lys430之间发生了阳离子-π 相互作用,其中吡啶基氮与P-loop残基Phe413和Gly414发生水介导的相互作用。将4-甲基吡啶-3-基用双环杂环取代,如在4L6中的吲唑(PDB:4z3V,图1-右)置换水分子并与P-Loop直接H-键相互作用,这导致发现了对BTK效力提高的配体如化合物4L6、以及1和2个 (见表1)。

Figure 1. Left: X-ray crystal structure of 4RV (PDB:4ZLZ) in the active site of Btk making a water mediated hydrogen bond with the P-loop backbone. Right: X-ray crystal structure of 4L6 (PDB:4Z3V) making direct H-bond interactions with the P-loop backbone.
在本算例中,我们使用了蛋白相互作用势和Flare提供的3D-RISM方法来研究BTK活性部位的静电和结晶水分子的稳定性。然后将该信息用于理解表1中分子的SAR。

方法
将4ZLZ和4Z3V配体-蛋白质复合物从蛋白质数据库下载到Flare中,并使用来自BioMolTech[5]的Build Model[4]工具小心地准备,以添加氢原子、优化氢键、消除原子冲突并将最佳质子化状态分配给蛋白质结构。任何截短的蛋白质链被封端作为蛋白质准备的一部分。
使用COBALT[6]多重比对工具在Flare中比对蛋白质序列,随后通过alpha碳原子的最小二乘拟合进行叠加。
蛋白的最小化
使用XED力场[7]和正常条件(梯度截断:0.200kcal/mol/ Å,最大迭代次数2,000次)使准备的4ZLZ和4Z3V配体-蛋白质复合物的活性位点在Flare中最小化。配体结构包括在活性位点的最小化中。
3D-RISM分析
参考相互作用位点模型(RISM)是一种基于分子Ornstein-Zernike方程的现代溶剂化方法[8] 。3D-RISM已经越来越多地用作研究蛋白质中水分子的位置和稳定性的方法。在概念上,3D-RISM等同于在溶剂上运行无限时间分子动力学模拟(保持溶质固定),然后提取溶剂颗粒的密度。 3D-RISM计算的输出包括含有颗粒密度的网格,一个用于氧,一个用于氢原子。热力学分析然后将ΔG值分配给网格上的每个位置,表示相对于大量水在网格的该位置处的假定水分子的”Happiness”。Flare中的3D-RISM计算使用了Cresset的XED力场,这提供了结合电子各向异性和一定程度的极化率的优点,从而提高了该方法的有效性。
在4ZLZ和4Z3V上进行了3D-RISM分析以研究Btk活性中心4RV和4L6配体周围结晶水分子的稳定性。使用了如下条件:
- XED force field and charge method
- 4Å grid spacing
- 14Å grid external border width
- Convergence tolerance: 10-8
- Maximum number of iterations: 10,000
- Total formal charge handling: neutralize with counterions.
蛋白相互作用势
蛋白相互作用势是Cresset分子相互作用势对蛋白质的延伸。两者都是使用XED力场计算的。该方法在原理上类似于配体场的计算:蛋白质的活性位点被探针原子充满,并且计算每个格点上的相互作用势。该方法利用了Mehler等人[9]的距离依赖的介电函数来更好地处理蛋白质结构中的大量带电基团。Table 1中的所有配体与4L6属于相同的系列,因此对于这种情况研究,仅计算和显示4Z3V的活性位点的蛋白质相互作用电势。
配体场
为了获得表1中配体的合理结合模式(pose),使用内置于Flare中的Lead Finder[10]方法将相应的2D结构对接到4Z3V的”干”(即,不包括结晶水分子)活性位点中。然后计算Cresset的配体场,并与4Z3V蛋白相互作用电势进行比较,以研究配体的SAR。然后计算Cresset的配体场,并与4Z3V蛋白相互作用电势进行比较,以研究配体的SAR。
结果
3D-RISM分析4ZLZ
在3D-RISM运行结束时,将3D-RISM水分子链添加到蛋白质结构中。该链中的水分子占据如3D-RISM所预测的高水密度区域,并且根据整个水分子计算的ΔG着色,在所有取向上取平均值。”Happy”水分子(ΔG为负值)被染成绿色:3D-RISM预测这些水分子在蛋白质中比在大量水中更稳定,因此更难被配体取代。”Unhappy”水分子(ΔG为正)呈红色:这些水相对于大量水不太稳定,因此更容易被配体置换。
图2显示了对4ZLZ进行3D-RISM计算的结果。氧密度表面(图2-左侧)清楚地显示了吡啶氮附近的局部水区域,3D-RISM局部算法(图2-右侧)表明水分子应该恰好存在于晶体结构中所见的位置。热力学分析表明,这种水分子既不是特别”happy”,也不是特别”Unhappy”。这与该水分子可置换的事实(如由4L6和表1中的其它化合物证明的)一致,但也表明置换基团需要具有正确的静电和形状以避免亲和力的损失。

Figure 2: 3D-RISM results on 4ZLZ. Left: oxygen isodensity surface at ρ=5. Right: localized 3D-RISM waters, colored by ΔG.
3D-RISM分析4Z3V
4Z3V的氧密度表面如图3-左侧所示。 3D-RISM定位算法正确地识别了结合到BTK活性部位的4L6配体周围的大部分晶体水分子的位置:预测到在这些水分子中有许多是”Happy”。因此,选定的一套稳定水分子的子集包含在4Z3V的蛋白质相互作用电势的计算中,因为它们被认为是配体与蛋白质活性位点结合的一部分。

Figure 3: 3D-RISM results on 4Z3V. Left: oxygen isodensity surface at ρ=5. Right: localized 3D-RISM waters, colored by ΔG.
4Z3V的蛋白相互作用势
如图4所示,4Z3V的”干”(不包括结晶水分子)和”湿”(包括排列在活性部位的稳定结晶型水分子)活性部位的蛋白质相互作用电势以令人满意的方式与4L6配体场匹配。尤其是:
- 富含电子的Cinnoline环位于4Z3V活性中心的正相互作用势区域;
- Cinnoline环的5,6-氢位于Leu408羰基的负相互作用静电势区域附近;
- 3-甲酰胺的羰基和NH2分别位于Met477的骨架NH和Btk的铰链区Glu475骨架羰基的正相互作用电势和负相互作用电势的区域内和附近,它们与该区域形成氢键;
- Cinnoline环上的4-氨基也位于负相互作用静电势的区域附近,对应于Met477和Leu408的羰基;
- 吲唑的富电子5元环位于Lys430的质子化侧链(未显示)和Phe413的骨架NH的正相互作用电势的区域中,其中NH基团指向Gly414骨架的负区域,Gly414与其形成氢键。
在计算蛋白质相互作用势时将稳定的水分子包含在内证实了这种情况。然而,在这种情况下,4Z3V活性部位中间的正蛋白相互作用势的区域大得多,并且将Cinnoline-吲唑环系统的大部分包含在内。这确实与Cinnoline-吲唑环系统周围的负配体场完全一致(图4-底部)。此外,Cinnoline环上的4-氨基位于负相互作用势的区域中,与正的配体场良好匹配。

Figure 4: 4L6 superimposed to the protein interaction potentials of 4Z3V. Top-left: ‘dry’ active site, not including crystallographic water molecules. Top-right: ‘wet’ active site including stable water molecules. Bottom: Ligand fields for 4L6. Protein interaction potentials shown at isolevel = 3; ligand fields shown at isolevel = 2.
BTK抑制剂的SAR
配体场与BTK活性位点的蛋白相互作用势的比较提供了对表1中化合物的SAR的一些有用的洞察。
Compound 1
化合物1(pIC50=8.7)是该数据系列中两个最有效的化合物之一[3],在吲唑环上带有-OMe侧链,在Cinnoline环的5位带有氟。化合物1的结合模式(图5)与4L6的结合模式相似。该化合物在铰链区与Glu475和Met477发生氢键相互作用,与Lys430(未显示)发生阳离子-π相互作用,以及与P-loop残基Phe413和Gly414的骨架发生氢键相互作用。氟基团位于相对大的口袋中,靠近一个可能被置换的水分子。 OMe基团中的CH3位于负相互作用势的区域中。

Figure 5: Left: compound 1 (pIC50 = 8.7) superimposed to the protein interaction potentials for the active site of 4Z3V at isolevel = 3. Right: ligand fields for compound 1 at isolevel = 2.
Compound 2
化合物2也是数据系列中最有活性的化合物之一。[3]然而,非常有趣的是,吲唑上的NH不与Gly414形成H键,因为它被转向了另一侧,可能与附近的水分子形成氢键相互作用。

Figure 6: Compound 2 (pIC50 = 8.7) superimposed to the protein interaction potentials for the active site of 4Z3V at isolevel = 3.
Compounds 3与4
化合物3的良好活性(pIC50=8.4)证实了双环系上的氢键供体不是BTK配体达到良好活性水平的必要特征。非常有趣的是,化合物4(pIC50=7.7)在结构上非常类似于3,但活性明显较低。如图7所示,比较这两个化合物的配位场与4Z3V活性部位的蛋白相互作用势可能为这个差异提供解释。而对这两个化合物(图7-中列),负配体场与Phe413的骨架NH的正相互作用势显示出良好的互补性,化合物4的正配体场(图7-右列)与Gly414骨架羰基产生的负相互作用势不匹配。在两个化合物Cinnoline环的7位上的甲基在确保配体在活性位点中实现正确构象方面与4L6吲唑环上甲基起到相同的作用。
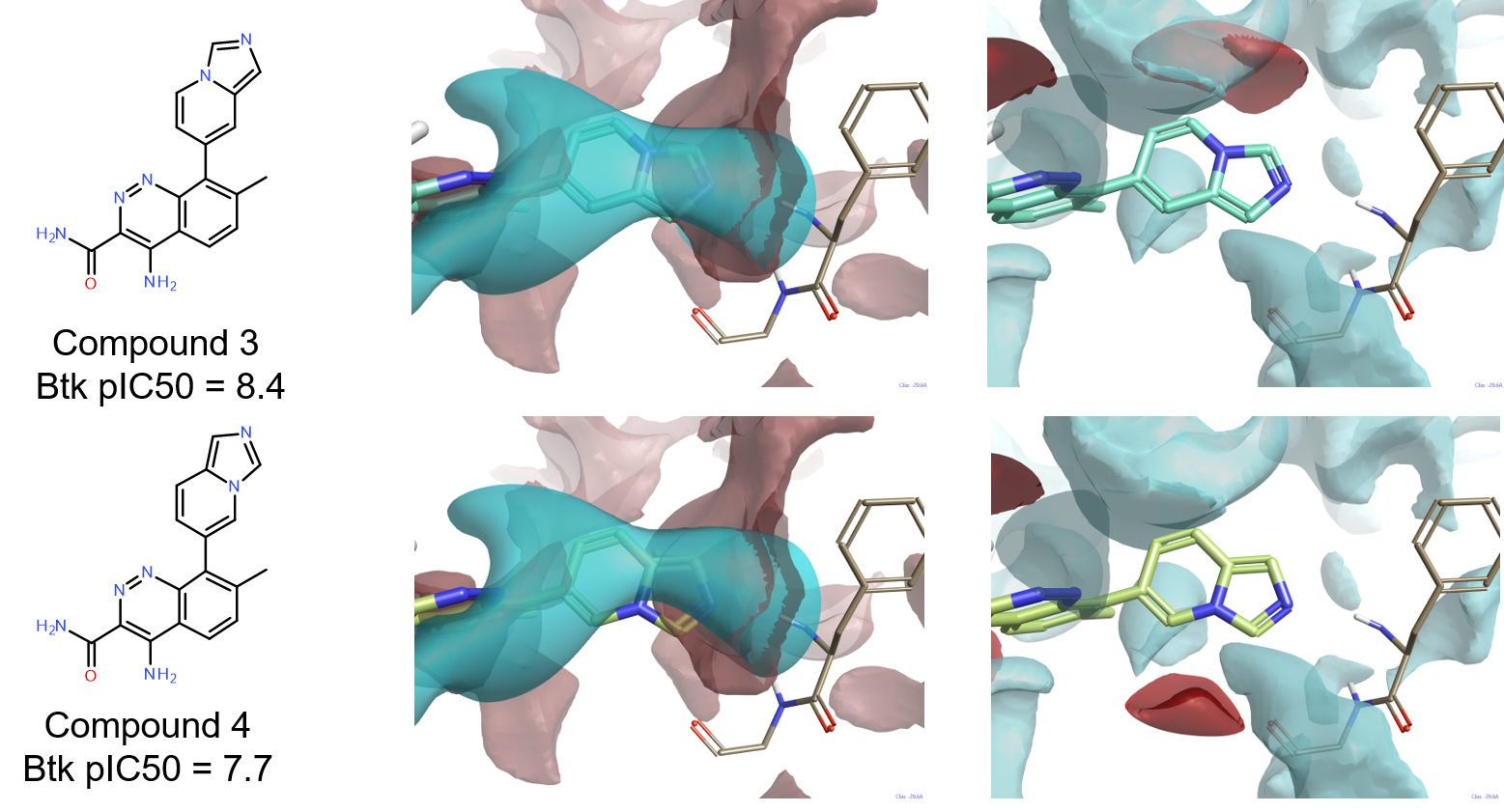
Figure 7: Compounds 3 and 4 superimposed to the protein interaction potentials for the active site of 4Z3V at isolevel = 3. Ligand fields shown at isolevel = 4.
Middle: positive interaction potentials superimposed to negative ligand fields.
Right: negative interaction potentials superimposed to positive ligand fields.
结论
在Flare中实现的蛋白相互作用势和配体场是理解配体-蛋白相互作用的静电的有力方法。在3D-RISM分析之后包含稳定的水分子显著提高了用于表征蛋白质活性位点的方法的精度。从蛋白质相互作用势获得的信息可用于指导配体设计,比较相关蛋白质以识别选择性机会,并从蛋白质的角度理解SAR趋势和配体结合。
文献
1. http://www.cresset-group.com/products/flare/
2. C.R. Smith et al., J. Med. Chem. 2015, 58, 5437−5444
3. US patent 2015/0038510
4. V. Stroganov et al., Proteins 2011, 79(9), 2693-2710
5. https://www.biomoltech.com/
6. https://www.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi
7. J.G. Vinter, J. Comput.-Aided Mol. Des. 1994, 8, 653-668
8. R. Skyner et. al., Phys. Chem. Chem. Phys. 2015, 17(9), 6174
9. E. L. Mehler, The Lorentz-Debye-Sack theory and dielectric screening of electrostatic effects in proteins and nucleic acids, in Molecular Electrostatic Potentials: Concepts and Applications, Theoretical and Computational Chemistry Vol. 3, 1996
10. O. V. Stroganov et al., J. Chem. Inf. Model. 2008, 48(12), 2371-2385