Flare V8发布
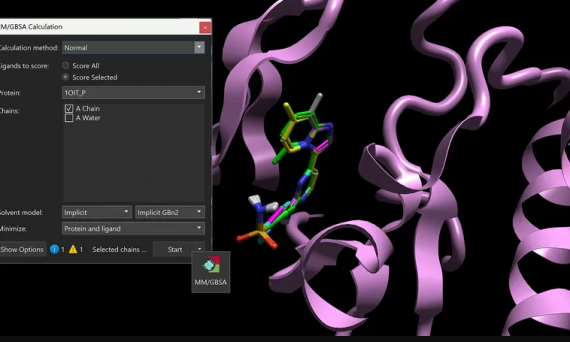
本文介绍了Cresset的CADD工作台Flare V8新增与增强的科学功能和方法,包括MM/GBSA方法用于计算配体-蛋白质复合物结合自由能,Flare FEP支持电荷变化,GIST水分析支持GCNCMC采样,HOMO/LUMO轨道的计算和显示,组合库枚举新增100多种反应,蛋白选项新增侧链构象生成功能等等。
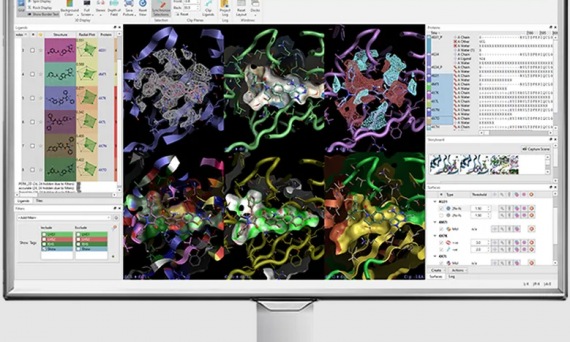
本文介绍了Cresset的CADD工作台Flare V8新增与增强的科学功能和方法,包括MM/GBSA方法用于计算配体-蛋白质复合物结合自由能,Flare FEP支持电荷变化,GIST水分析支持GCNCMC采样,HOMO/LUMO轨道的计算和显示,组合库枚举新增100多种反应,蛋白选项新增侧链构象生成功能等等。
本文介绍了最新版本Flare V5的功能与特性,包括:整合了基于配体的方法Forge;基于结构与基于配体方法的协同使用;GIST水分子分析方法;增强的FEP相对结合自由能计算;更快速的分子动力学模拟与增强的结果分析表单;基于场的药效团模拟与生物活性构象预测;全功能Python API。
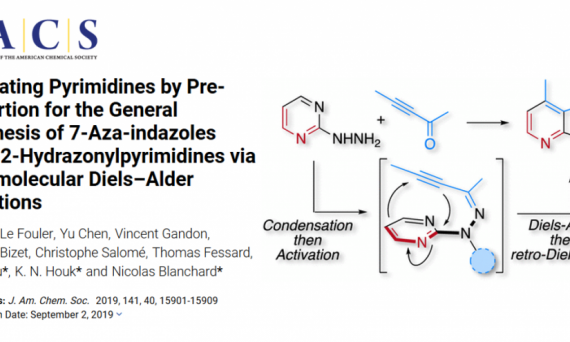
目前大多数计算研究还局限在理解现有的实验现象上,将计算化学作为一个手段,在研究初期提供定量的指导还并不多见,为了探究这种可能性,我们介绍了一篇2019年Blanchard课题组发表在JACS上的工作,我们将采用一种问题导向的策略逐步趋向目标的实现,并阐明如何利用理论计算实现一个反应的设计:嘧啶与炔烃的环加成反应。
由Jame B. Foresman & Eleen Frisch 合著,徐雪飞翻译的《探索化学的奥秘:电子结构方法》第三版是一本Gaussian软件入门指南,旨在介绍如何应用电子结构方法模拟化合物和反应,适合于没有经验的实验研究化学家、物理化学专业的高年级本科生与初级研究生、Gaussian软件用户使用。
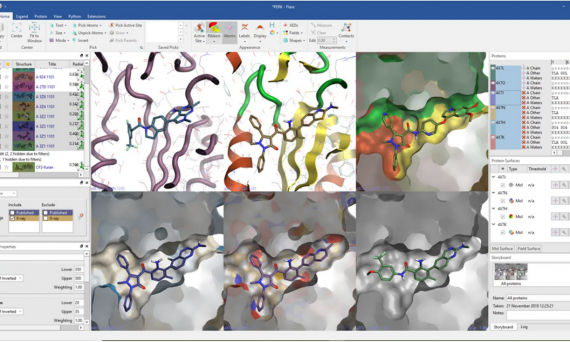
利用强大的分子可视化技术和Cresset久经考验的静电技术可以深入了解蛋白质-配体复合物结构,通过高质量的图形和交互式分析可以有效地传达您的想法。Flare Viewer是基于结构药物设计软件Flare的免费许可组件,它是一款先进的分子可视化工具。在Flare Viewer中您可以使用免费的组件。
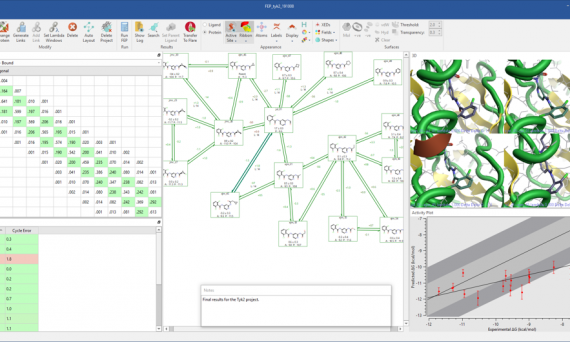
FLARE V3正式发布,新特性包括可靠、充分验证的自由能微扰(FEP)计算;多种分子对接(Docking)方式,包括共价对接、模板对接,提升性能的Lead Finder分子对接;整合了Forge的基于配体的叠合,可以进行构象搜索、基于场、形状与子结构叠合;还提供了分子动力学模拟以及100多个新功能与改进。
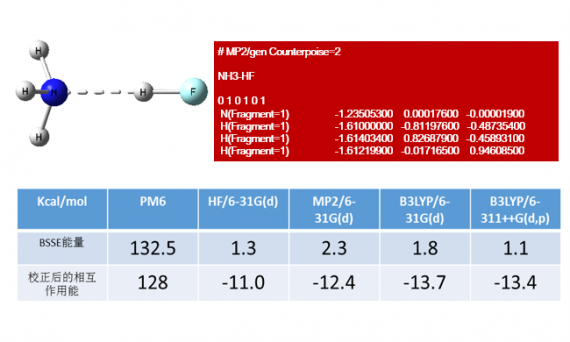
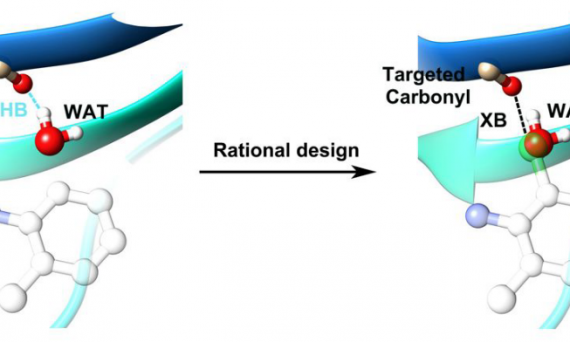
本文介绍了氢键(H-Bond)、卤键(X-Bond)、分子间相互作用能以及基组重叠误差(BSSE)等相关概念。并以HF与NH3形成的氢键和IF与NH3形成的卤键为例,详细演示了如何利用Gaussian程序中的CP校正方法修正基组重叠误差(BSSE),以获得更精确的分子间相互作用能。
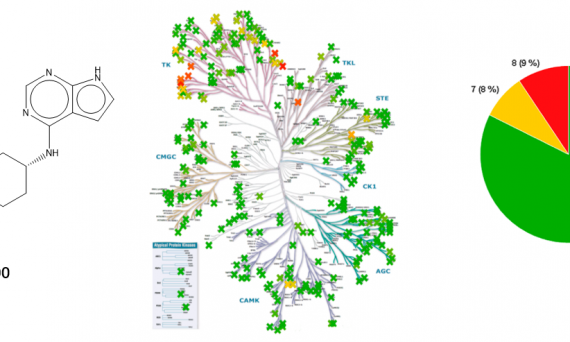
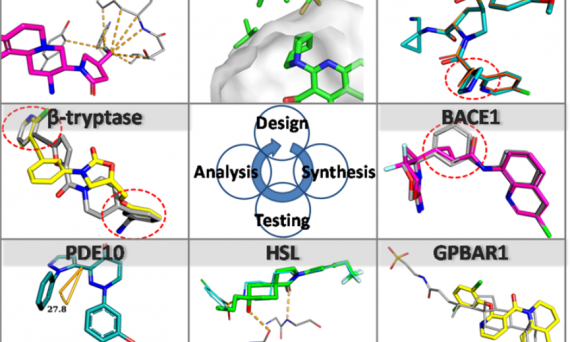
大量致力于JAK激酶抑制剂的发现工作最终将几种化合物推进到临床开发并有两个被FDA批准。尽管在过去20年中付出了巨大的努力,但是高选择性的JAK3抑制剂一直未能找到。辉瑞公司的研究人员经过巨大的努力发现了首个口服活性的JAK3特异性抑制剂,其通过与JAK3特有的残基CYS-909共价相互作用实现JAK同工酶选择性抑制。JAK3酶共价抑制剂设计的难点在于其具有相对快速的再合成速率,因此要求该酶的共价抑制剂不仅具有合适的药动学性质(pharmacodynamics properties)而且还要将不希望的脱靶反应性限制到最小范围。本研究的努力最终发现了化合物11(PF-06651600),不仅具有体内活性而且清除率较低。鉴于化合物11的有利药效和安全性,该化合物进入临床研究评价阶段。