AutoDock教程 (ChemBio3D版)
一、背景介绍
Tyrosine-protein kinase ABL1是白血病的一个重要靶点,比如格列卫就是作用于这个靶点。本教程以格列卫(STI)衍生物与靶点复合物晶体结构出发,演示如何用ChemBio3D为AutoDock界面进行分子对接计算。
二、准备工作
1,安装ChemBio3D Ultra 14或ChemBioOffice Ultra 14
为商业软件需要购买或安装试用版:http://www.molcalx.com.cn/chembio3d-ultra-14
2,安装AutoDockTools
点击Calculations>>AutoDock Interface>>Install AutoDock/AutoDockTools,按照提示安装。
3,安装AutoDock4.2
点击Calculations>>AutoDock Interface>>Install AutoDock/AutoDockTools,按照提示安装。
4,下载受体与配体文件,记录AutoDock Grid map中心坐标
1)登陆结合位点数据库:http://www.molcalx.com.cn/targetdb
2)在query检索框输入”1fpu”,选1fpu那行,点击search
3)下载现成的mol2配体文件以及受体pdbqt文件
4)解压,获得ligand.mol2(配体文件),protein.pdbqt(受体文件)共两个文件
5)在AutoDock Search space区给出结合位点中心坐标,可以作为对接的Grid map中心坐标
center_x = 13.0974
center_y = 96.9759
center_z = 57.9442
三、操作步骤
1,打开AutoDock Interface
点击Calculations>>AutoDock Interface>>Setup AutoDock Calculation
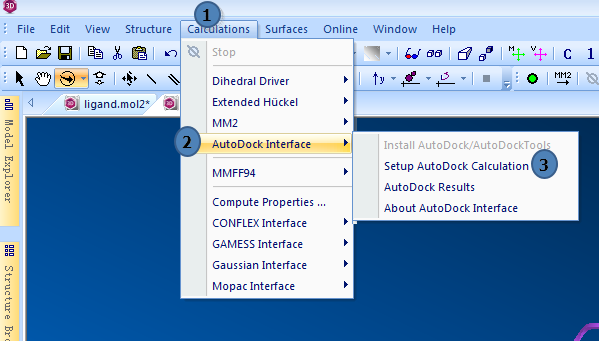
图1,打开AutoDock Interface
2,准备受体
点击AutoDock Interface>>Prepare receptor,按图2操作。
第(5)步,读入之前准备好的受体文件,protein.pdbqt。
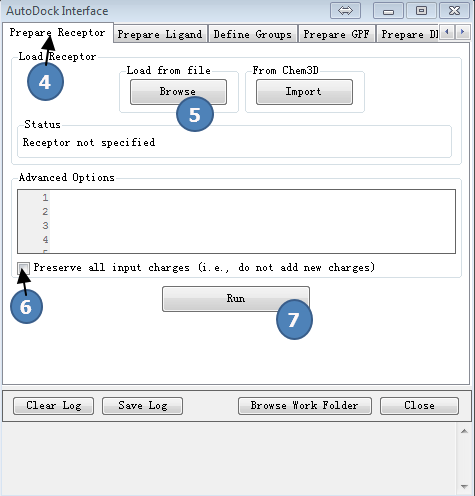
图2,准备受体,载入预先准备好的受体文件,最后点Run按钮
3,准备配体
点击AutoDock Interface>>Prepare Ligand
第(9)步,读入之前准备好的配体文件,ligand.mol2。
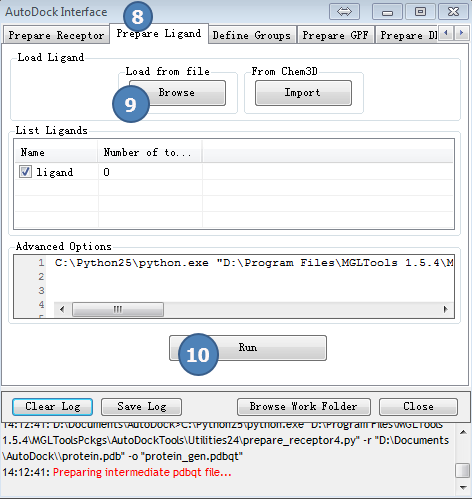
图3,准备配体,载入预先准备好的配体文件,ligand.mol2, 最后点Run按钮
4,准备GPF
返回AutoDock Interface>>Prepare GPF
输入GRID Map中心坐标(来自准备工作,从结合位点数据库获的AutoDock search space处获取信息):
X:13.7
Y:97
Z:58
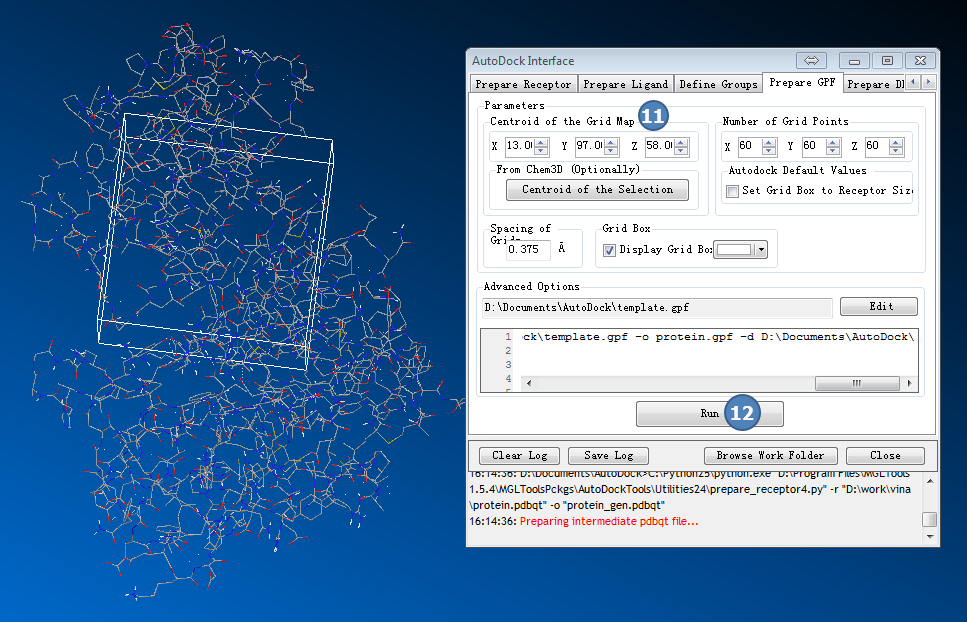
图4,准备GPF,填入结合位点数据库给的结合位点中心坐标,点Run按钮, 成功后会有个盒子出现。
5,准备DPF
点击AutoDock Interface>>Prepare GPF
采用预设值,点“Run”按钮。
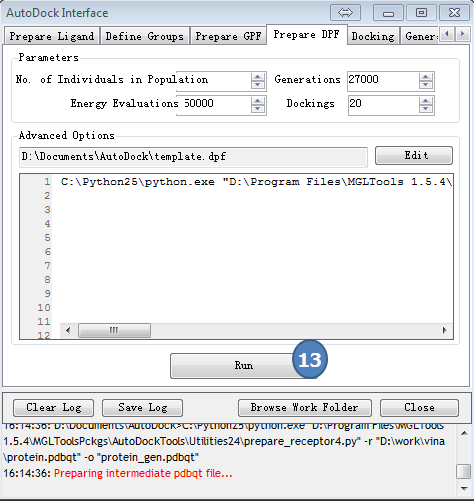
图5,准备DPF,采用预设值,点Run按钮。
6,Docking计算
点击AutoDock Interface>>Docking
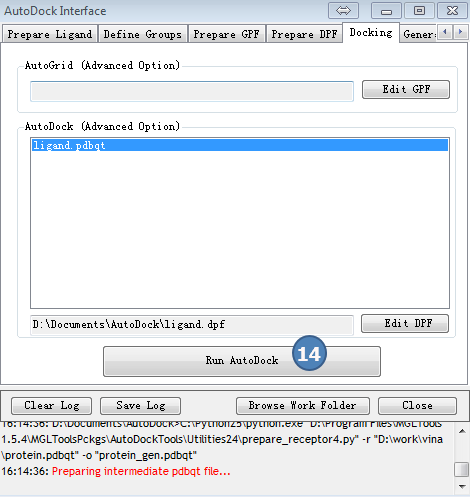
图6,docking,采用预设值,点Run AutoDock按钮。